Saúde
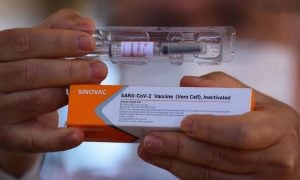
Ministério da Saúde afirma que comprará toda a produção da Coronavac
Segundo a pasta, as doses serão incorporadas ao Plano Nacional de Operacionalização da Vacinação contra a Covid-19


O Ministério da Saúde informou neste sábado 9 que todas as doses da Coronavac produzidas pelo Instituto Butantan, de São Paulo, serão compradas com exclusividade pelo governo federal. Segundo a pasta, as doses serão incorporadas ao Plano Nacional de Operacionalização da Vacinação contra a Covid-19.
A decisão, segundo o ministério, foi acertada em uma reunião na sexta-feira 8 entre representantes da pasta e do instituto. Nos próximos dias, haverá uma reunião entre o ministro Pazuello e representantes dos secretários estaduais e municipais de saúde para detalhamento da logística de distribuição e do calendário da campanha de vacinação.
Em nota, o Ministério da Saúde afirmou que as doses da vacina serão distribuídas aos estados em quantidade proporcional à população de cada um e que os estados farão a distribuição do imunizante para os municípios.
Diretor do Butantan confirma operação
Neste sábado, o diretor-geral do Instituto Butantan, Dimas Covas, reconheceu a incorporação de todas as doses da vacina Coronavac pelo governo federal.
“No momento, o Butantan tem 6 milhões de doses e serão incorporadas pelo ministério, a medida que houver liberação do uso emergencial. Essas vacinas serão distribuídas por todos os estados de forma proporcional, obedecendo critérios demográficos e número de pessoas nas faixas de risco. Todos os estados serão atendidos. Com essas e mais 2 milhões vindo pela FioCruz. Teremos em janeiro 8 milhões de doses para iniciar a campanha [nacional] de vacinação”, afirmou.
Ainda de acordo com o executivo do Butantan, a expectativa é que a distribuição da vacina comece em até 48 horas da liberação do uso emergencial feita pela Anvisa.
Em nota, o instituto explicou que os contratos firmados com o Ministério da Saúde possuem cláusula de exclusividade” e “a partir do momento em que o Butantan fornece as doses ao órgão federal, cabe ao Programa Nacional de Imunizações distribuí-las aos estados, conforme os quantitativos solicitados pelas Secretarias de Saúde, inclusive a de São Paulo”.
O Butantan explicou que serão fornecidas 46 milhões de doses ao Ministério da Saúde. Na quantidade, estão inclusas as 10,8 milhões que foram adquiridas da biofarmacêutica Sinovac e já estão em solo brasileiro.
A negociação, ainda de acordo com o instituto, não impacta a negociação para o fornecimento da vacina a outros países.
Anvisa diz que faltam dados do Butantan para uso emergencial
Também neste sábado, a Anvisa informou, em nota, que a documentação apresentada pelo Instituto Butantan para liberação do uso emergencial da Coronavac está incompleta.
“A submissão dos documentos técnicos previstos no Guia é condição necessária para viabilizar a avaliação, conclusão e a deliberação sobre a autorização de uso emergencial das vacinas”, diz trecho da nota.
Entre as informações adicionais que a Anvisa solicitou ao Butantan estão dados de “características demográficas e basais críticas da população do estudo (idade, sexo, raça, peso ou IMC) e outras características (por exemplo, função renal ou hepática, comorbidades)” e a contabilização de “dados sobre a disposição dos participantes, com uma contabilidade clara de todos os participantes que entraram no estudo”.
A Anvisa afirmou que já está em contato com o Butantan para discutir “prazos e cronogramas para apresentação dos dados faltantes”. Neste sábado, as equipes técnicas da agência e do instituto já realizaram duas reuniões para tratar da questão.
A agência também informou que no caso da Fiocruz os documentos estão completos e a análise do pedido segue para a próxima fase.
Veja na íntegra a nota da Anvisa sobre o Butantan
Neste sábado (9/1/202), a Anvisa concluiu a triagem dos documentos submetidos pelo Instituto Butantan para autorização de uso emergencial da vacina CoronaVac.
Esses documentos haviam sido recebidos na manhã de ontem, sexta-feira. Às 22 horas de ontem, a Anvisa recebeu do Instituto informações adicionais, dentro do mesmo processo.
Após a triagem de todos os documentos fornecidos, os técnicos da Anvisa verificaram que ainda faltam dados necessários à avaliação da autorização de uso emergencial.
A partir da triagem, a Anvisa enviou hoje, sábado, 09/01, ofício ao Instituto Butantan solicitando a apresentação dos documentos técnicos faltantes, previstos no Guia 42/2020 (Requisitos para submissão de solicitação de autorização temporária de uso emergencial Vacinas – COVID-19), bem como nos regulamentos técnicos da Agência. O recebimento do ofício foi confirmado pelo Butantan às 11h29 de hoje.
A submissão dos documentos técnicos previstos no Guia é condição necessária para viabilizar a avaliação, conclusão e a deliberação sobre a autorização de uso emergencial das vacinas.
No dia de hoje, sábado, as equipes técnicas da Anvisa e do Instituto Butantan já realizaram duas reuniões tratar da questão.
O Instituto foi informado sobre a necessidade dos documentos complementares, essenciais à análise e conclusão sobre a eficácia e segurança da vacina. Na oportunidade, foram discutidos prazos e cronogramas para a apresentação dos dados faltantes.
A checagem é uma conferência, uma triagem inicial, feita nas primeiras 24 horas para verificar se as informações essenciais sobre eficácia e resultados clínicos estão no processo para análise de uso emergencial pela equipe técnica da Anvisa.
São essas as seguintes informações e resultados que ainda deverão ser apresentados:
1. Características demográficas e basais críticas da população do estudo (idade, sexo, raça, peso ou IMC) e outras características (por exemplo, função renal ou hepática, comorbidades). Essas características demográficas e basais críticas devem ser apresentadas por braços do estudo e tipo de população de análise “intenção-de-tratamento” (ITT) e “por protocolo”(PP), de forma a permitir a comparabilidade dos grupos de tratamento.
2. Resultados do estudo por população de “intenção-de-tratamento” (ITT).
3. Dados sobre a disposição dos participantes, com uma contabilidade clara de todos os participantes que entraram no estudo. O número de pacientes que foram randomizados e que entraram e completaram cada fase do estudo (ou cada semana/mês do estudo) devem ser fornecidos, bem como as razões para todas as interrupções pós-randomização, agrupados por tratamento e por motivo principal (perda de acompanhamento, evento adverso, pobre conformidade, etc.).
4. Descrição dos desvios de protocolo ocorridos no estudo com a adequada classificação de impacto e de categoria.
5. Listagem de participantes com desvios de protocolo, divididos por centro.
6. Dados de imunogenicidade do estudo fase 3.
Porque é importante a Anvisa analisar essas informações?
As informações são essenciais para a confiabilidade do estudo apresentado. O grau de confiança nos resultados gerados por um estudo clínico, também chamado de validade interna, deve ser avaliado por uma autoridade sanitária para permitir concluir pela eficácia e segurança de uma vacina experimental. A validade interna de um estudo clínico é o grau em que os resultados obtidos refletem os verdadeiros resultados dos estudos e, portanto, não seriam devidos a erros metodológicos. A validade interna de um ensaio clínico está diretamente relacionada ao delineamento, condução e relatos apropriados do estudo clínico.
O Instituto Butantan informou que apresentará os dados com brevidade e a Anvisa continuará a avaliar a documentação que já foi enviada, de forma a otimizar esforços para uma decisão célere sobre o pedido.
Adicionalmente, a Anvisa esclarece que seguirá com a análise de todos os documentos já submetidos, de modo a agilizar o máximo possível o processo de avaliação e autorização de vacinas COVID-19. Além disso, os dados já avaliados pela Anvisa submetidos pelo procedimento de submissão contínua não precisarão ser reanalisados pela agência.
Veja a íntegra da nota da Anvisa sobre a FioCruz
Neste sábado (9/1/2021), a Anvisa concluiu a triagem inicial dos documentos submetidos pela Fundação Oswaldo Cruz (Fiocruz) para autorização de uso emergencial da vacina de Oxford.
De acordo com a triagem feita pelos técnicos da Anvisa, o pedido traz os documentos preliminares e essenciais para a avaliação detalhada da Agência.
A partir de agora, a equipe técnica vai se aprofundar na análise dos dados e informações apresentadas pela Fiocruz.
A checagem é uma conferência, uma triagem inicial, feita nas primeiras 24 horas para verificar se as informações essenciais sobre eficácia e resultados clínicos estão no processo para análise de uso emergencial pela equipe técnica da Anvisa.
Para a avaliação do uso emergencial, a Anvisa também utiliza os dados que já haviam sido submetidos pelo laboratório por meio da Submissão Contínua, ou seja, pacotes de dados prontos enviados anteriormente enquanto outras etapas da pesquisa seguiam em andamento.




CartaCapital
Há 31 anos, a maior referência progressista em jornalismo no Brasil.
Apoie o jornalismo que chama as coisas pelo nome
Muita gente esqueceu o que escreveu, disse ou defendeu. Nós não. O compromisso de CartaCapital com os princípios do bom jornalismo permanece o mesmo.
O combate à desigualdade nos importa. A denúncia das injustiças importa. Importa uma democracia digna do nome. Importa o apego à verdade factual e a honestidade.
Estamos aqui, há mais de 30 anos, porque nos importamos. Como nossos fiéis leitores, CartaCapital segue atenta.
Se o bom jornalismo também importa para você, nos ajude a seguir lutando. Assine a edição semanal de CartaCapital ou contribua com o quanto puder.